From 1 January 2021 a new regime will apply to medical devices placed on the market in the UK. New medical device regulations for the UK were published in 2019.
Those regulations essentially copy and paste the MDR and IVDR into UK law, albeit that this would be as a parallel system to that in the EU, unless mutual recognition is agreed. However new guidance from the MHRA suggests that these regulations might be repealed and replaced by an entirely new regulatory regime unique to the UK. A consultation in "autumn" is promised, but time is running short.
In the interim, the most recent MHRA guidance informs us of a new regime for the UK which treats Northern Ireland differently from Great Britain (England, Scotland and Wales). This is necessary because of the terms of the Northern Ireland Protocol agreed with the EU. That protocol avoids separating the island of Ireland along the border between the Republic of Ireland and Northern Ireland. This brings added complexity for medical device manufacturers because of different labelling requirements in Great Britain and Northern Ireland.
CE marked devices
Medical device manufacturers who have CE marked devices (whether under the EU Directives or the EU Regulations) will be able to continue to place them on the market in the whole of the UK until 1 July 2023 without a change in labelling. After that, devices destined for Great Britain will be required to follow the UK regulatory regime and to be labelled with the UKCA mark. Northern Ireland will, however, continue to accept CE marked devices.
Even if using the CE mark in the UK after 1 January 2021, there are some additional hurdles for manufacturers.
Appointing a UK Responsible Person
The first hurdle is that manufacturers who are based outside the UK, whether in the EU, the EEA or elsewhere, will need to appoint a UK Responsible Person (UKRP). The UKRP will take on regulatory responsibilities with respect to the MHRA and users or customers in the UK which include:
- verifying that regulatory requirements have been met, retaining and then providing regulatory documentation to the MHRA on request
- interacting with the MHRA on regulatory matters and acting as a go-between with the manufacturer
- keeping the manufacturer informed about complaints and reports of suspected incidents, and
- terminating the legal relationship with the manufacturer if the latter breaches its regulatory obligations and informing the MHRA of that termination.
Registration requirements
The second hurdle to overcome to allow devices to be placed on the market in the UK after 1 January 2021 (even for CE marked devices), is registration. A UK manufacturer must register with the MHRA, as must a UKRP for an overseas manufacturer. The registering entity will then register each of the devices for which they are responsible for placing on the market in the UK, whether in Great Britain or Northern Ireland.
The following grace periods (from 1 January 2021) apply to the requirements both to appoint a UKRP and to register the devices:
- Four months: Class IIIs and Class IIb implantables, and all active implantable medical devices and IVD List A
- Eight months: Class IIb non-implantable medical devices, and all Class IIa devices, IVD List B and self-test IVDs
- 12 months (unless currently required to register with the MHRA): Class I devices and general IVDs.
We assume that even if IVDs are placed on the UK market under the IVD Regulations (as opposed to the Directive), that the grace period will be determined according to the classification set out in the Directive.
However, for general medical devices that are up classified or newly regulated devices under MDR, it is not clear which rules would apply. Of course, the MDR will never be fully applicable in the UK, but we assume that the classification under MDR would only be applied from 26 May 2021, unless the new UK legislation says otherwise.
UKCA mark and Northern Ireland mark (UKNI)
Manufacturers who are not supplying the EU/EEA markets might prefer the UK regulatory system to the EU's. Additionally, all device manufacturers will need to start to transition to the UK system in advance of the deadline of 30 June 2023. We have very little information on that new system except for the following:
- There will be UK Approved Bodies (equivalent to EU notified bodies) and notified bodies previously authorised under the EU Directives will automatically qualify for this role.
- There is to be a new UKCA mark to be added to a device that conforms with the UK regulatory system and which cannot be used in the EU, EEA or (on its own) in Northern Ireland.
- Devices solely destined for the market in Northern Ireland and following the UK authorisation process must be labelled with the UKNI mark, which similarly cannot be used on devices destined for the market in the EU or EEA but can be combined with the UKCA mark in Northern Ireland.
- Manufacturers following the EU regulatory system and who have an Authorised Representative in Northern Ireland and are placing devices on the market in Northern Ireland (not Great Britain) need not appoint a UK Responsible Person: the Authorised Representative in Northern Ireland can instead fulfil this role regarding the MHRA obligations.
Any manufacturer using the UKCA mark must from 1 January 2021 include the name and address of the UK Responsible Person and the number of the Approved Body on the label. Additionally, where in Northern Ireland any importer is neither the manufacturer nor the Authorised Representative, then the UKRP or Northern Ireland Authorised Representative must also provide the importer's details to the MHRA.
UK Manufacturers supplying in the EU/EEA
UK manufacturers selling their devices into the EU/EEA will not be able to include the UKCA mark or UKNI mark on these devices. For the purpose of CE marking, they will, of course, require a notified body in the EU/EEA although not in Northern Ireland, - none exist there in any event. UK manufacturers, including those based in Northern Ireland will require an authorised representative, but they can be based in the EU, EEA or even in Northern Ireland.
Supervision and enforcement
Vigilance reporting will continue to be made to the MHRA for the whole of the UK, and the MHRA will continue to be the enforcement authority for the UK. The MHRA will not though be able to challenge certifications, decisions or authorisations emanating from EEA regulatory bodies, whether notified bodies or competent authorities in relation to devices placed on the market in Northern Ireland.
It will be able to apply restrictions to any devices placed on the market in Great Britain, even if they are of the same type as those placed on the market in Northern Ireland because the acceptance of CE marked products in Great Britain is at the UK government's discretion. The MHRA will nevertheless have enforcement powers in relation to devices placed on the market in Great Britain and/or Northern Ireland and which do not meet the requirements of EU law.
For a summary of the requirements by location of manufacturer and where the device is placed on the market, please see the table below.
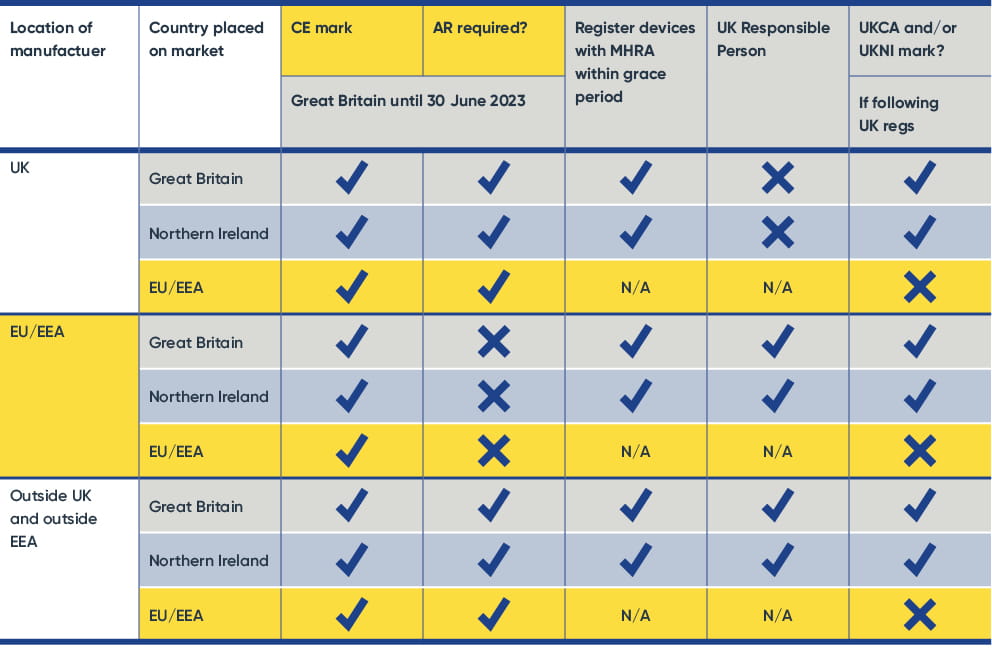
Please note:
- AR can be in EU, EEA or Northern Ireland.
- You cannot combine a CE mark with UKCA and/or UKNI marks for devices to be placed on the market in the EU or EEA – CE mark only there; you can combine CE mark with UKCA and/or UKNI mark for devices destined for Great Britain.
- Northern Ireland requires the UKNI mark, which can be combined with the UKCA mark.
Find out more
To discuss the issues raised in this article in more detail, please contact a member of our Life Sciences and Healthcare team.

How to prepare for the MHRA's new medical devices enforcement rules
Key actions you can take to prepare for the new enforcement rules.
1 / 6 观点

Patent pools: an easy licensing option for COVID-19 drugs and SARS CoV 2 vaccines?
An exploration of patent pools, R&D and current initiatives.
2 / 6 观点

Private equity investment in life sciences sector
Exploring some of the potential benefits and challenges for life sciences companies considering PE investment.
4 / 6 观点

Targeted treatments – Part 4: patenting digital devices
Important advances are being driven by digital communications technology, particularly in the medical devices sector.
5 / 6 观点

UK increases scrutiny of tech and life sciences deals
Mergers and acquisitions in the tech and life sciences sectors may be subject to closer scrutiny by the UK competition authority and by the UK government in future.
6 / 6 观点
返回